I’m working on revisions for a paper and I honestly need to get this task done and so for today I will link enhancers to genes and run motif discovery analysis.
Terminology
There are 23 pairs of chromosomes in humans and collectively the DNA is over 3 billion base pairs in length.
Think of DNA as a string of length >3 billion.
Some regions of the DNA are genes, a functional unit passed from parent to child determining characteristics such as eye colour.
Genes are regions on the DNA.
And other regions of the DNA are enhancers, which can be bounded by proteins to increase the likelihood of a gene being activated.
Enhancers are also regions on the DNA.
Task 1: Linking enhancers to nearby genes
The enhancer and gene input files are in BED format.
Format: BED
Tab separated with the following columns:# Required 1. chrom - the name of the chromosome 2. chromStart - index of region start 3. chromEnd - index of region end # Optional 4. name 5. score 6. strand
We link enhancers to genes using Bedtools (specifically the “window” command).
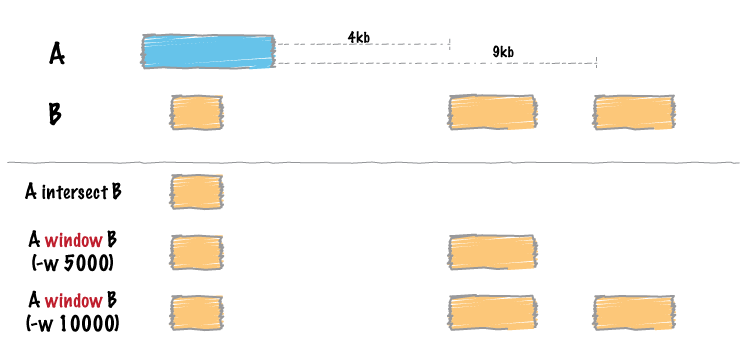
Note: In the above figure from Bedtools, the blue (in my situation) represents a gene of interest and the orange represents the enhancers.
Running the following command will give us the subset of enhancers near the genes of interest:
bedtools window -a ENHANCER.bed -b GENE.bed -w 1000000 -u
In this command, option “-u” is used to print original entry A (i.e. enhancers) found in B (i.e. genes); with a window “-w” of 1 Mbp.
We can then run motif analysis on the enhancers of interest.
Task 2: Motif discovery
We use HOMER for motif discovery.
The file input format to HOMER is a BED file with 4 columns (see above).
# Generate HOMER input file directly from Task 1
$output_of_task1 | awk -F'\t' '{print $1, $2, $3, $1":"$2"-"$3}' OFS="\t"
We run HOMER as follows:
# Make sure HOMER/bin and WebLogo is in PATH environment
HOMER/bin/findMotifsGenome.pl $HOMER_INPUT_BEDFILE hg19 $OUTPUT_DIR -size given
We now wait for the jobs to be done.
A bit more complicated
Underneath the hood, it’s a bit more complicated than what I described. For instance, my analysis is in R and so my intervals are stored as GRanges objects to which I have to output into BED files.
library(GenomicRanges)
library(dplyr)
write_grranges_to_bed <- function(genes_of_interest, output_bedfile){
ensemblgtf[ensemblgtf$gene_id %in% genes_of_interest] %>%
{
data.frame(
chrom = seqnames(.),
chromStart = start(.),
chromEnd = end(.),
geneId = mcols(.)$gene_id,
strand = strand(.),
stringsAsFactors = FALSE)
} %>%
group_by(geneId, strand) %>%
summarise(
chrom = sprintf("chr%s", unique(chrom)),
chromStart = min(chromStart),
chromEnd = max(chromEnd)
) %>%
ungroup() %>%
mutate(score = "1") %>%
select(chrom, chromStart, chromEnd, geneId, score, strand) %>%
arrange(chrom, chromStart) %>%
readr::write_tsv(output_bedfile, col_names = FALSE)
}
I also (fortunately) have a make pipeline setup so that I can use a Makefile to run HOMER:
HOMER ?= $(SOFTWARE_DIR)/HOMER/v4.8
WEBLOGO ?= $(SOFTWARE_DIR)/weblogo/v2.8.2
findMotifsGenome = $(HOMER)/bin/findMotifsGenome.pl
build ?= hg19
homer_opt ?= -size given
$(homer-outdir)%: $(homer-input-file)
$(eval posfile = $<)
$(eval outdir = $@)
export PATH=$(HOMER)/bin:$(WEBLOGO):$$PATH ;\
$(findMotifsGenome) $(posfile) $(build) $(outdir) $(homer_opt)